Excitotoxicity מאופיינת כעלבון חריף הגורם למוות של תאי עצב עקב הפעלה מוגזמת של iGluRs. רעילות חריפה ממלאת תפקיד מהותי במגוון בעיות בריאותיות של מערכת העצבים המרכזית (CNS), כולל איסכמיה מוחית, TBI ומצב אפילפטי. המנגנונים לרעילות חריפה שונים עבור כל נושא בריאותי. �
עם איסכמיה במוח, אקציטורעילות הקשורות ל-L-גלוטמט ו-L-אספרטאט מתרחשות תוך דקות עקב הצמיחה של L-גלוטמט מוחי חוץ-תאי וכן L-אספרטאט. מכיוון שאלו תלויות גם באנרגיה, אובדן פתאומי של אנרגיה עקב הפסקת זרימת הדם יכול בסופו של דבר לפרק את הממברנה העצבית והאסטרוגליאלית. בנוירונים, דה-פולריזציה של הממברנה תורמת לפריקה שלפוחית. בנוסף, פירוק אנרגיה עלול אפילו לגרום לשינוי בפעולתם, ולכן, לגרום ל-L-גלוטמט ו-L-אספרטאט להפעיל ולהשפיע על הומאוסטזיס יוני שעלול להפריע לפעולת EAAT. ההפעלה של L-glutamate/L-aspartate תורמת ל-exitotoxicity באמצעות הפעלת יתר של iGluRs מסוג NMDA כפי שהוכחה על ידי היעילות של אנטגוניסטים NMDA במודלים של בעלי חיים של איסכמיה מוחית חולפת. �
ב-TBI, הנזק המכני לרקמות והשיבוש של מחסום הדם-מוח עלולים לעורר ניוון נוירו-מוח משני חריף, אשר יחד עם דלקת עצבית ולחץ חמצוני, קשור להפעלת L-גלוטמט מתאים תוך-תאיים, ולכן, לרעילות חריפה. מעבר לכך, יישום חריף של האנטגוניסט NMDA MK801 בעקבות TBI משפר בין היתר אובדן נוירוני והפרעות התנהגותיות ארוכות טווח. �
בסטטוס אפילפטיקוס, המשך הפעילות המסונכרנת של רשתות נוירונים מעוררות כמו גם פירוק מתמשך של מנגנונים מגבילים הוא המקור העיקרי להפעלת L-glutamate ו-L-aspartate. מכיוון שחומרת הפעילות הסינכרונית תלויה במעורבות של תאי עצב במערכת עצבית וכן ביכולתו של תא עצבי לעמוד בעודף גלוטמט תלויה בעיקר בדפוס הביטוי של iGluRs, ניוון מעט מוגבל וקשור להתבגרות של אוכלוסיות עצביות. אשר בסופו של דבר נגרמת מהתקפים אפילפטיים ממושכים. המשמעות של אקסיטוטוקסיות בסטטוס אפילפטיקוס מוצגת כאנטגוניסטים של NMDA, כגון קטמין, מפחיתים את אובדן האדרנל. �
אקסיטוטוקסיות במחלות נוירולוגיות
מכיוון ש-EAATs התגלו כמווסתות מטה במגוון בעיות בריאותיות של מערכת העצבים המרכזית (CNS) ו-L-גלוטמט, כמו גם L-aspartate, פינוי יכול בסופו של דבר להשפיע על האקסיטוטוקסיות של מחלות נוירולוגיות, אנשי מקצוע רבים בתחום הבריאות החליטו לקבוע חומרים הגורמים ל-EAAT2, או ל-EAAT העיקרי במוח, ובדרך כלל הוכח כבעלי ירידה בוויסות. זה הוכיח חומרים שמראים ביטוי אסטרוציטי של EAAT2 הן במחקר מבחנה והן במחקר in vivo. כמה מהם הוכיחו גם תכונות הגנה במודלים של בעלי חיים של מחלות נוירולוגיות. Cef היא אחת התרכובות המוערכות ביותר והיא נותחה במודלים של AD, HD ו-ALS עם תוצאות חיוביות. עם זאת, אף אחד מהחומרים לא נחקר בהרחבה בשל יכולתו לקיים אינטראקציה עם מסלולים נוירו-פרוקטיבים אחרים. כמו כן, הוכח כי Cef מקדם את ביטוי EAAT2 אך גם מעורר את גורם השעתוק Nrf2, מה שגורם לשעתוק של מגוון רחב של גנים המעורבים בהגנה על ציטו-פרוטקציה והגנה על נוגדי חמצון. מכיוון שמאמינים שלחץ חמצוני ממלא תפקיד חיוני ברבות, אם לא כולן, מחלות נוירולוגיות, מסלול זה עשוי להסביר את ההגנה העצבית הנגרמת על ידי Cef. יתר על כן, הוכח כי xCT, שיכול להיות אחד מהמטרות במורד הזרם של Nrf2, מווסת על ידי Cef in vitro ו-in vivo. חומר נוסף המקדם EAAT2 במבחנה, MS-153, מוגן ביעילות מפני ניוון עצבי משני לאחר פציעה מוחית טראומטית וכן באמצעות מנגנונים אחרים מלבד ויסות EAAT2. עדויות לניסויי קונספט המדגימים את הגירוי המוגבר באמצעות iGluRs במחלות נוירודגנרטיביות זקוקות למניפולציות של הפיזיולוגיה של הנוירוטרנסמיטר שלהם. �
עכברי Glud1 Tg מדגימים מודל של אקסיטוטוקסיות הקשורה להפעלה מוגברת של L-גלוטמט סינפטית עם אובדן נוירוני מוגבל. עם זאת, מודל החיות הזה של העברה עצבית גלוטמטרית עדיין לא נוצל כדי לנתח אם ביטוי יתר של Glud1 מחמיר את הפנוטיפ של מודלים של עכברים במחלות נוירולוגיות. גרסה אחרת כוללת את העכבר החסר EAAT2. לעכברי נוק-אאוט הומוזיגוטים EAAT2 יש בעיות בריאות הקשורות למוות בטרם עת בגלל אפילפסיה וכן ניוון בהיפוקמפוס ובקליפת המוח המוקדית. עכברי נוק-אאוט הטרוזיגוטיים EAAT2, לעומת זאת, מתפתחים באופן תקין ומציגים הפרעות התנהגותיות קלות בלבד. מודל עכבר זה של תפקוד יתר של גלוטמט מתון נוצל באוסף של עדויות למחקרים עקרוניים שהדגימו את התפקיד הבסיסי של גלוטמט. עכברי ALS, שיש להם גם את המוטציה G93A mSOD1 וגם כמות מופחתת של EAAT2 (SOD1(G93A)/EAAT2�), חשפו עלייה במהירות הירידה המוטורית מלווה באובדן נוירון מוטורי מוקדם יותר בהשוואה לעכברי G93A mSOD1 Tg בודדים. . ירידה בהישרדות הודגמה גם בעכברים מוטנטים אלה. כאשר מצטלבים עם עכברים מהונדסים המבטאים מוטציות של העמילואיד האנושי-? מבשר חלבון ופרסנילין-1 (A?PPswe/PS1?E9), אובדן חלקי של חסרונות זיכרון מרחביים חשופים של EAAT2 בעכברים בני 6 חודשים המבטאים A?PPswe/PS1?E9. עכברים אלה הדגימו עלייה ביחס של A?42/A?40 שאינו מסיס בדטרגנט, והוכיחו כי מחסור בתפקוד טרנספורטר גלוטמט גורם בסופו של דבר לתהליכים פתוגניים מוקדמים הקשורים ל-AD. לשם השוואה, הפנוטיפ של מודל העכבר R6/2 HD לא השתנה בעכברים שהיו להם רק אלל EAAT2 אחד. מחקרים נוספים עדיין נחוצים להוכחות נוספות. �
כהשלמה למחקרים אלו, פותחו גם עכברים מהונדסים המבטאים יתר על המידה EAAT2 באסטרוציטים באמצעות מקדם GFAP. עכברי EAAT2/G93A mSOD1 Tg כפולים הפגינו שיפור מתון של הפנוטיפ דמוי ה-ALS שלהם עם עיכוב מובהק סטטיסטית (פי 14) בירידה בכוח האחיזה ואובדן של נוירונים מוטוריים, כמו גם ירידה בהזדמנויות אחרות, כגון הפעלת קספאז-3 ו SOD1, אם כי לא בתחילתו של שיתוק, ירידה במשקל או תוחלת חיים ממושכת בהשוואה לבני חולי G93A mSOD1 חד-טרנסגניים. בדיוק אותו מודל עכבר מהונדס EAAT2 נוצל כדי להעריך את ההשפעה של ספיגת L-גלוטמט אסטרוציטית ו-L-אספרטאט משופרת על ידי הכלאה עם מודל חיה של עכברי AD, A?PPswe/Ind. רמות מוגברות של חלבון EAAT2 העלו ושיפרו באופן ניכר את התפקוד הקוגניטיבי הכללי, החזירו את האתיקה הסינפטית והקטינו את הפלאק העמילואיד באותם עכברי AD. �
בעכברים שבהם ויסות וניהול של xCT מהונדסים גנטית גורמים לחוסר במערכת האנטי-פורטר של גלוטמט/ציסטין x?c, הירידה הברורה של L-גלוטמט אקסטרה-סינפטי קשורה לעמידות האדירה של נוירונים דופמינרגיים נגד ניוון עצבי המושרה על ידי 6-הידרוקסידופמין. אולי כתוצאה מהפחתת הרעילות המעוררת. עם זאת, הוכח גם כי הפעלת מיקרוגליה מווסתת על ידי ליקויים במערכת x?c המובילים לפנוטיפ נוירו-פרוטקטיבי יותר המציע הסבר להשפעה המגנה של מחיקת xCT בנסיבות אלה. �
לכן, וריאציות גנטיות מעודדות את התפקיד של אקזיטוקסיות כרונית במחלות נוירודגנרטיביות, במיוחד AD ו-ALS. מודלים אלה מייצגים כולם שינויים לכל החיים בהולכה עצבית גלוטמטרגית. מודלים אלה אינם יכולים לקבוע אם השימוש בתרופות ו/או תרופות יכול להשפיע ישירות על רמות הגלוטמט לאורך התהליך הנוירו-דגנרטיבי ו/או להגן. הן הערכה והן ניתוח של רפואה מעוררת EAAT2 להתקדמות של מודלים של עכברים הניתנים להשראה והאינטראקציה שלהם עם מסלולי איתות אחרים עדיין מוצדקות על ידי חוקרים ואנשי מקצוע בתחום הבריאות. �
במחקרי מחקר רבים, הוכחות ומדדי תוצאות הראו כי הפרעה ברמת הגלוטמט וההתרגשות במחלות נוירולוגיות רבות, כולל AD, HD ו- ALS, מביאים בסופו של דבר להתנוונות עצבית ולמגוון הסימפטומים הקשורים לבעיות הבריאותיות. מטרת המאמר שלפניכם היא לדון ולהדגים את התפקיד שמסולדת הפרעת הגלוטמט וההתרגשות על מחלות עצביות. המנגנונים להרעיית יתר שונים בכל נושא הבריאותי. - ד"ר אלכס חימנז DC, CCST Insight - ד"ר אלכס Jimenez DC, תובנה CCST
טופס הערכה מטבולית
ניתן למלא את טופס ההערכה המטבולי הבא ולהגיש בפני ד"ר אלכס חימנז. קבוצות סימפטומים המופיעות בטופס זה אינן מיועדות לשמש כאבחון לכל סוג של מחלה, מצב או סוג אחר של בריאות.
אקסיטוטוקסיות מאופיינת כעלבון חריף הגורם למוות של תאים עקב הפעלת עודף של iGluRs. לטיפול במערכת העיכול ממלא תפקיד מהותי במגוון סוגיות בריאותיות של מערכת העצבים המרכזית (CNS), כולל איסכמיה מוחית, TBI ואפילפטיקוס סטטוס. המנגנונים להרעיית חריפה הם שונים בכל נושא הבריאותי. היקף המידע שלנו מוגבל לנושאי כירופרקטיקה, שרירים ושלד עצבים וכן מאמרים, נושאים ודיונים ברפואה תפקודית. אנו משתמשים בפרוטוקולים בריאותיים תפקודיים לטיפול בפציעות או בהפרעות כרוניות במערכת השלד והשרירים. לדיון נוסף בנושא לעיל, אנא אל תהסס לשאול את ד"ר אלכס חימנז או צור איתנו קשר בכתובת 915-850-0900 .
אוצר על ידי ד"ר אלכס חימנז
הפניות
לברנץ, יאן ופמלה מאהר. רעילות גלוטמט כרונית במחלות ניווניות - מהי הראיות? גבולות בתחום מדעי המוח, Frontiers Media SA, 16 דצמבר 2015, www.ncbi.nlm.nih.gov/pmc/articles/PMC4679930/.
דיון בנושא נוסף: כאב כרוני
כאב פתאומי הוא תגובה טבעית של מערכת העצבים המסייעת להפגנת פגיעה אפשרית. דרך אגב, אותות כאב נודדים מאזור פצוע דרך העצבים וחוט השדרה למוח. בדרך כלל הכאב פחות חמור ככל שהפציעה מחלימה, עם זאת, כאבים כרוניים שונים מהסוג הממוצע של הכאב. עם כאבים כרוניים, גוף האדם ימשיך לשלוח אותות כאב למוח, ללא קשר אם הפגיעה נרפאה. כאב כרוני יכול להימשך מספר שבועות ואף מספר שנים. כאבים כרוניים יכולים להשפיע בצורה אדירה על ניידות המטופל והוא יכול להפחית את הגמישות, הכוח והסיבולת.
צוהר עצבי פלוס למחלות נוירולוגיות
ד"ר אלכס חימנז משתמש בסדרת בדיקות המסייעות להערכת מחלות נוירולוגיות. הזומר העצביTM בנוסף הוא מערך של נוגדנים אוטומטיים נוירולוגיים המציע זיהוי נוגדנים-לאנטיגן ספציפי. הזומר העצבי התוססTM פלוס נועד להעריך את תגובתו של האדם ל- 48 אנטיגנים נוירולוגיים עם קשר למגוון מחלות הקשורות לנוירולוגיה. הזום העצבני התוססTM Plus שואפת להפחית מצבים נוירולוגיים על ידי העצמת חולים ורופאים עם משאב חיוני לגילוי סיכונים מוקדם והתמקדות משופרת במניעה ראשונית בהתאמה אישית.
נוסחאות לתמיכה במתילציה
XYMOGEN s נוסחאות מקצועיות בלעדיות זמינות באמצעות אנשי מקצוע נבחרים בתחום הבריאות. מכירה והנחה באינטרנט של נוסחאות XYMOGEN אסורות בהחלט.
בגאווה, ד"ר אלכסנדר חימנז עושה XYMOGEN נוסחאות זמין רק לחולים תחת הטיפול שלנו.
נא להתקשר למשרד שלנו על מנת שנוכל להקצות רופא התייעצות לגישה מיידית.
אם אתה חולה של פגיעה רפואית וכירופרקטיקה קלינית, אתה יכול לשאול על XYMOGEN על ידי קורא 915-850-0900.
מחקרים קודמים מצביעים על כך ש-L-אספרטאט, כמו L-גלוטמט, מעורר פעילות מעוררת על נוירונים. L-aspartate מתפקד עם L-glutamate בשלפוחיות הסינפטיות של סינפסות מעוררות אסימטריות. אבל, הריכוז הכולל של אלה במוח האנושי (0.96-1.62 ?מול/גרם משקל רטוב), הריכוזים החוץ-תאיים שלהם בקליפת המוח כפי שנמדד במיקרודיאליזה (1.62 ?M ל-L-aspartate ו-9.06?M ל-L-גלוטמט) והאספקה שלהם על פי אימונוהיסטוכימיה מצביעות על כך ש-L-אספרטאט נמצא בכמות נמוכה משמעותית מ-L-גלוטמט. יתר על כן, L-aspartate הוא אגוניסט רב עוצמה עבור קולטני NMDA אך לא עבור iGluRs אחרים עם EC50 גבוה רק פי שמונה מזה של L-glutamate. EAATs אשר ממלאים תפקיד מהותי בספיגת כל ה-L-גלוטמט המשוחרר שלפוחית במערכת העצבים המרכזית (CNS) דורשים גם ניצול של L-aspartate. L-אספרטאט הוא אולי פחות חיוני כמו L-גלוטמט המחובר לפעילות המעוררת הכוללת הקשורה ל-iGluRs. לצד תפקידו כנוירוטרנסמיטר, כאמור, L-aspartate נחוץ גם כמצע ל-aspartate amino-transferase אשר הופך ל-2-oxoglutarate ו-L-glutamate להובלה לשלפוחיות הקורטיקליות של נוירונים גלוטמטריים אשר עשויות גם כתוצאה מכך להגביר בעקיפין את שחרור L-גלוטמט. �
מולקולות אחרות באיתות גלוטמט
מאפיין אחד המבדיל בין קולטני NMDA לבין iGluRs שונים הוא שהפעלת קולטני NMDA מצריכה חיבור של קו-אגוניסט לאזור קושר הגליצין של הקולטן. לדוגמה, ברשתית ובחוט השדרה, המקור של גליצין עלול לזלוג מתוך סינפסות מעכבות גליצינרגיות. אבל, באזורים שונים של המוח עם ביטוי מוגבר של קולטן NMDA, כגון היווצרות ההיפוקמפוס, חסרות תגובות הקשורות לקולטני גליצין רגישים לסטריכנין, לפחות בנוירונים בוגרים, המדגימות את היעדר העברות עצביות מעכבות גליצינרגיות. עם זאת, גליצין נמצא בנוזל החוץ-תאי של ההיפוקמפוס בכמויות בסיסיות של בערך 1.5 ?M, אשר דומה לרוויה של אזור קושר הגליצין של קולטן ה-NMDA, אם כי אלה עשויים להיות מווסתים למעלה ולמטה. מקורו של גליצין חוץ תאי בהיפוקמפוס יכול להיות נוירונים המשחררים גליצין דרך טרנספורטר חומצות האמינו אלנין-סרין-ציסטאין 1 (asc-1). אבל, הוכח גם שחרור גליצין על ידי אסטרוציטים המומר על ידי דה-פולריזציה וקאינט. מחקרים נוספים נדרשים כדי להראות בסופו של דבר מדדי תוצאה אלה. �
אפילו במחקרים קודמים של קולטן ה-NMDA והפעלתו המשותפת על ידי גליצין, גילו כי חומצות אמינו D, במיוחד D-serine, חזקות כמעט כמו גליצין. רק מספר שנים לאחר מכן, התברר ש-D-serine נמצא במוחות של חולדות ובני אדם בערך בשליש מריכוז ה-L-serine שלהם עם ריכוז מוחלט של יותר מ-0.2 μmol/g רקמת מוח. תוך שימוש באנטי-סרום ל-D-serine, מחקרים הראו ש-D-serine מהמוח נמצא רק באסטרוציטים והאספקה שלו מתאימה לביטוי של קולטני NMDA. בנוסף, אותם חוקרים הדגימו ש-D-serine משתחרר מאסטרוציטים מתורבתים כאשר הם נחשפים ל-L-גלוטמט או קיינט. השפע של D-serine נמצא על ידי האנזים המשפיל D-amino acid oxidase (DAO) החושף ביטוי מוגבר במוח האחורי שבו רמות ה-D-serine מופחתות וכן האנזים הסינתטי serine racemase אשר יוצר D-serine מ-L- סרין. נראה כי D-Serine מאוחסן בשלפוחיות ציטופלזמיות באסטרוציטים והוא יכול להשתחרר על ידי אקסוציטוזיס. התעצמות ארוכת טווח תלויה בשחרור D-serine מאסטרוציטים בפרוסות ההיפוקמפוס, מה שמרמז כי חומצת אמינו זו בהחלט משחקת תפקיד מהותי בהולכה עצבית גלוטמטרגית דרך קולטני NMDA. בנוסף בפרוסות ההיפוקמפוס, מחקרים מצאו, שימוש באנזימים מפרקי D-serine וגליצין, אשר D-serine מתפקד כמשדר משותף לקולטני NMDA סינפטיים על נוירונים CA1, כמו כן, גליצין מתפקד כקו-אגוניסט אנדוגני לקולטני NMDA חוץ-סינפטיים. קולטני NMDA סינפטיים של נוירוני גירוס דנטאטים משתמשים בגליצין ולא ב-D-serine בתור אגוניסט. �
נלקחים ביחד, מדדי תוצאה רב-שכבתיים מראים ש-L-אספרטאט לא פשוט מתפקד כאגוניסט על קולטני NMDA, אלא גם לגליצין ו-D-סרין תפקידים בסיסיים בהולכה עצבית גלוטמטרית במוח האנושי. אבל, מולקולות אחרות הוכחו גם כמאפננים רלוונטיים של העברה עצבית גלוטמטרגית. �
גלוטמט מופעל על ידי מולקולות אחרות
ל-L-homocysteate (L-HCA) יש קווי דמיון מבניים עם L-glutamate. חומצת האמינו שאינה חלבונית היא תוצר חמצון של הומוציסטאין שיוצר ביו-סינתזה מתיונין בסילוק קבוצת המתיל הסופנית שלה והיא גם תוצר ביניים של מסלול הטרנססולפורציה שבאמצעותו מתיונין עשוי להפוך לציסטאין באמצעות ציסטתיונין. מחקרים מוקדמים הוכיחו כי חומצת אמינו זו יכולה לגרום לזרימת סידן לנוירונים מתורבתים בצורה בטוחה ויעילה כמו L-גלוטמט. יתרה מכך, L-HCA חשף זיקה מוגברת לקולטני NMDA בהשוואה ל-iGluRs אחרים במבחני קשירה הקשורים ליכולתו לגרום ל-exitotoxicity הניתנת לעיכוב של אנטגוניסט של NMDA ולזרימת נתרן. בנוסף, L-HCA יכול להפעיל את mGluR5 ביעילות כמו L-גלוטמט. L-HCA נמצא במוח, עם זאת, הוכח כי הריכוזים היו נמוכים בערך פי 500 מאלה של L-גלוטמט ואפילו פי 100 פחות בהשוואה לאלו של L-aspartate באזורים שונים של מוח החולדות. במהלך הגירוי המושרה על ידי אשלגן, פריקת L-HCA מופעלת מתכשירי פרוסות מוח כפי שהוכח עבור L-aspartate ו-L-glutamate, אם כי השחרור המוחלט של HCA קטן פי 50 בערך. באופן מפתיע, HCA הוא מעכב תחרותי יעיל מאוד של ספיגת ציסטין ו-L-גלוטמט דרך מערכת אנטי-פורטר ציסטין/גלוטמט x?c, הפעילות המווסתת ומנהלת את ריכוזי ה-L-גלוטמט החוץ-תאיים במוח. לכן, ההשפעה של L-HCA על ההפעלה של NMDA וקולטני L-גלוטמט אחרים עשויה להסתמך גם על הטריגר המושרה על ידי L-HCA של L-גלוטמט דרך מערכת x?c. L-HCA עשוי לשחק תפקיד חשוב בגירוי הכולל של קולטני L-גלוטמט. עם זאת, זה יכול להשתנות מאוד בתנאים מסוימים, למשל בחולים עם טיפול במינון גבוה של מתוטרקסט, תרופה אנטי סרטנית אשר, על ידי הגבלת דיהידרופולאט רדוקטאז, מגבילה את המיחזור המזוזז של מתיונין מהומוציסטאין. כאן, ריכוזי L-HCA של יותר מ-100 ?M הוכחו מנוזל השדרה בעוד ש-L-HCA לא היה ניתן לגילוי בנבדקי ביקורת. מחקרים נוספים נדרשים עדיין כדי לקבוע מדדי תוצאה אלה. �
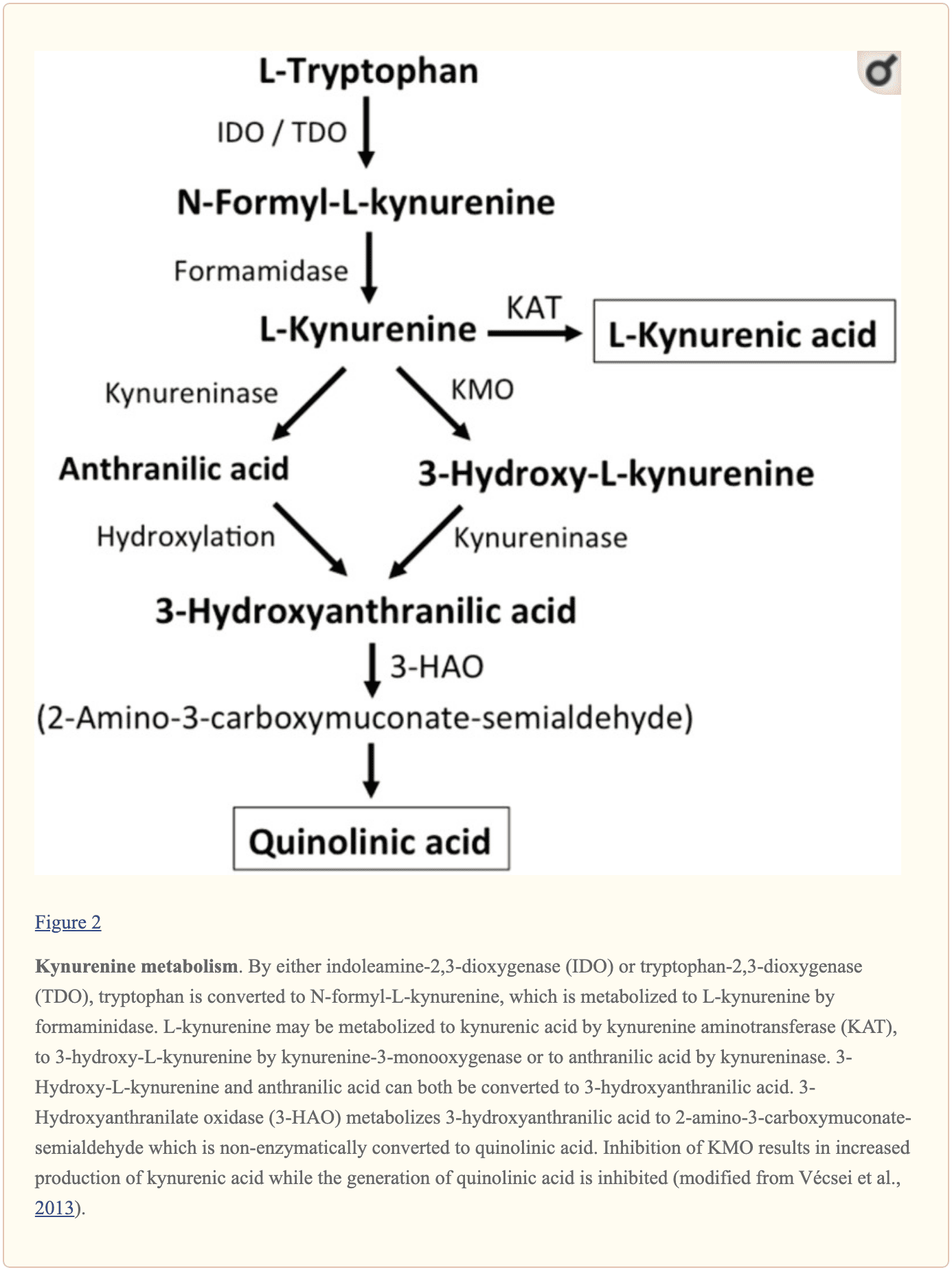
מולקולות קטנות אנדוגניות נוספות אשר מאמינים כי הן משפיעות על איתות L-גלוטמט כוללות מספר תוצרי ביניים של מטבוליזם טריפטופן, כפי שמוצג באיור 2. באמצעות הפעילות של אינדולאמין 2,3-דיאוקסיגנאז (IDO) או טריפטופן 2,3-דיאוקסיגנאז (TDO), טריפטופן הופך ל-N-formyl-L-kynurenine אשר הופך מאוחר יותר ל-kynurenine (KYN) על ידי פורמידאז. שלושה מסלולים, שניים מהם מתחברים בשלב הבא, מביאים לחילוף חומרים נוסף. ראשית, באמצעות הפעילות של kynurenine aminotransferase (KAT), KYN הופך לחומצה kynורנית (KYNA). ניתן להמיר KYN גם ל-3-hydroxykynurenine (3HK) על ידי kynurenine monooxygenase (KMO), אשר ניתן לאחר מכן לנצל כמצע על ידי kynureninase לסינתזה של חומצה 3-hydroxyanthranilic (3HANA). בנוסף, תוך שימוש ב-KYN כמצע, kynureninase מפתח חומצה אנתרנילית (ANA), אשר על ידי הידרוקסילציה לא ספציפית עשויה להיות מומרת ל-3HANA. על פי מחקרים, 3HANA מתפקדת סוף סוף כמצע לייצור חומצה כינולינית (QUIN). �
ריכוז הטריפטופן במוח החולד הוא בערך 25 ננומול/גרם משקל רטוב וכפי 400 פחות מ-L-גלוטמט ופי 100 פחות מ-L-אספרטאט. רמות המוח שהוכחו של kynurenines נמוכות עוד יותר עם 0.4-1.6 ננומול/ג עבור QUIN, 0.01-0.07 ננומול/מ"ל עבור KYNA ו-0.016 ננומול/ג עבור 3HANA. כ-40 אחוז מה-KYN במוח מסונתז באופן מקומי. המטבוליטים של טריפטופן מדגימים קישור דיפרנציאלי לחלבוני פלזמה והובלתם דרך המחסום שהוא שונה בתכלית. KYN ו-3HK נישאים דרך מערכת נושאת חומצות אמינו נייטרליות גדולות L. Kynurenines כאילו חודרים למוח האנושי על ידי דיפוזיה פסיבית. בנוסף, KYNA, 3HANA, ובמיוחד ANA נקשרות לחלבוני סרום אשר בסופו של דבר מגבילים ומגבילים את יכולת הפיזור שלהם על פני מחסום הדם-מוח. �
מחקרים הוכיחו ש-QUIN, כאשר נעשה שימוש יונופורטי בתאי חולדה, גרם לירי נוירוני אשר נמנע על ידי אנטגוניסט לקולטן NMDA, מה שמצביע על כך ש-QUIN עשוי לתפקד כאגוניסט לקולטן NMDA. עם זאת, הוכח כי ה-EC50 עבור QUIN להפעלת זרמי קולטן NMDA גבוה פי 1000 בערך מה-EC50 של L-גלוטמט. הוכחה שהזרקה תוך מוחית של QUIN גורמת לשינויים אולטרה-מבניים, נוירוכימיים והתנהגותיים הדומים לאלו הנגרמים על ידי אגוניסטים לקולטן NMDA. העובדה שריכוזי QUIN נמוכים בערך פי 5000 עד פי 15,000 מריכוזי L-גלוטמט במוח הופכת את זה לא סביר שאפנון איתות קולטן NMDA על ידי QUIN משחק תפקיד חיוני. הוכח כי KYNA מתפקד כאנטגוניסט לקולטן NMDA. אבל, למרות עירוי עם מעכב ה-KMO Ro 61-8048 שיפרה את ריכוזי ה-KYNA החוץ-תאיים המוחיים פי 10, זה לא הביא לעיכוב של דה-פולריזציה עצבית בתיווך NMDA, ממצא שמאתגר את האמונה ש-KYNA בכמויות כמעט פיזיולוגיות. מווסת את קולטני NMDA. לשם השוואה, KYNA מוגבר במוח המושרה ממעכבי KMO JM6 הפחית את ריכוז ה-L-גלוטמט החוץ-תאי. בנוסף, רמות KYNA מנוזל המוח החוץ-תאי נקשרו לרמות L-גלוטמט, דבר המצביע על כך שגם ברמות פיזיולוגיות או כמעט פיזיולוגיות, KYNA מווסת את חילוף החומרים של L-גלוטמט. הן ההפעלה של הקולטן המזוהה עם GPR35 והן העיכוב של קולטני ?7 ניקוטיני אצטילכולין פרה-סינפטיים מוצעים בהפחתה המושרה על ידי KYNA בשחרור L-גלוטמט. לסיכום, למרות ש-QUIN ו-L-HCA נמצאים במוח האנושי, הריכוזים שלהם דנים נגדם עם תפקידים בוויסות ותחזוקת העברה עצבית. לעומת זאת, למרות שיש להגדיר את המסלולים בפירוט רב יותר, עדויות תומכות ברמות ובדעה שניתן לווסת פריקה על ידי KYNA והולכה עצבית. �
גלוטמט, יחד עם מולקולות אספרטט ומולקולות אחרות, הם כמה מהמועברים העצביים העיקריים במוח האנושי. למרות שאלו ממלאים תפקיד בסיסי במבנה הכולל ובתפקוד מערכת העצבים המרכזית, כולל המוח וחוט השדרה, כמויות מופרזות של מולקולות אחרות יכולות בסופו של דבר לעורר קולטני גלוטמט. עודף גלוטמט יכול לגרום לרגשות רגישים שעלולים להוביל למגוון בעיות בריאותיות, כמו מחלת אלצהיימר וסוגים אחרים של מחלות נוירולוגיות. המאמר שלהלן מתאר כיצד מולקולות אחרות יכולות להפעיל קולטני גלוטמט. - ד"ר אלכס חימנז DC, CCST Insight - ד"ר אלכס Jimenez DC, תובנה CCST
מחקרים מראים כי L-aspartate, כמו L-glutamate, מעורר פעילות מעוררת. L-aspartate מתפקד עם L-glutamate בכוריות הסינפטיות של סינפסות מעוררות אסימטריות. עם זאת, הריכוז הכולל של אלה במוח האנושי מרמז על כך ש- L-aspartate הוא פחות פחות בשפע מאשר L- גלוטמט. יתר על כן, L-aspartate הוא אגוניסט רב עוצמה עבור קולטני ה- NMDA אך לא עבור iGluRs אחרים עם EC50 הגבוה פי שמונה מזה של L-glutamate. היקף המידע שלנו מוגבל לנושאי כירופרקטיקה, שרירים ושלד עצבים וכן מאמרים, נושאים ודיונים ברפואה תפקודית. אנו משתמשים בפרוטוקולים בריאותיים תפקודיים לטיפול בפציעות או בהפרעות כרוניות במערכת השלד והשרירים. לדיון נוסף בנושא לעיל, אנא אל תהסס לשאול את ד"ר אלכס חימנז או צור איתנו קשר בכתובת 915-850-0900 .
אוצר על ידי ד"ר אלכס חימנז
הפניות
לברנץ, יאן ופמלה מאהר. רעילות גלוטמט כרונית במחלות ניווניות - מהי הראיות? גבולות בתחום מדעי המוח, Frontiers Media SA, 16 דצמבר 2015, www.ncbi.nlm.nih.gov/pmc/articles/PMC4679930/.
דיון בנושא נוסף: כאב כרוני
כאב פתאומי הוא תגובה טבעית של מערכת העצבים המסייעת להפגנת פגיעה אפשרית. דרך אגב, אותות כאב נודדים מאזור פצוע דרך העצבים וחוט השדרה למוח. בדרך כלל הכאב פחות חמור ככל שהפציעה מחלימה, עם זאת, כאבים כרוניים שונים מהסוג הממוצע של הכאב. עם כאבים כרוניים, גוף האדם ימשיך לשלוח אותות כאב למוח, ללא קשר אם הפגיעה נרפאה. כאב כרוני יכול להימשך מספר שבועות ואף מספר שנים. כאבים כרוניים יכולים להשפיע בצורה אדירה על ניידות המטופל והוא יכול להפחית את הגמישות, הכוח והסיבולת.
צוהר עצבי פלוס למחלות נוירולוגיות
ד"ר אלכס חימנז משתמש בסדרת בדיקות המסייעות להערכת מחלות נוירולוגיות. הזומר העצביTM בנוסף הוא מערך של נוגדנים אוטומטיים נוירולוגיים המציע זיהוי נוגדנים-לאנטיגן ספציפי. הזומר העצבי התוססTM פלוס נועד להעריך את תגובתו של האדם ל- 48 אנטיגנים נוירולוגיים עם קשר למגוון מחלות הקשורות לנוירולוגיה. הזום העצבני התוססTM Plus שואפת להפחית מצבים נוירולוגיים על ידי העצמת חולים ורופאים עם משאב חיוני לגילוי סיכונים מוקדם והתמקדות משופרת במניעה ראשונית בהתאמה אישית.
נוסחאות לתמיכה במתילציה
XYMOGEN s נוסחאות מקצועיות בלעדיות זמינות באמצעות אנשי מקצוע מורשים בתחום הבריאות. המכירה וההנחה באינטרנט של נוסחאות XYMOGEN אסורות בהחלט
בגאווה, ד"ר אלכסנדר חימנז עושה XYMOGEN נוסחאות זמין רק לחולים תחת הטיפול שלנו.
נא להתקשר למשרד שלנו על מנת שנוכל להקצות רופא התייעצות לגישה מיידית.
אם אתה חולה של פגיעה רפואית וכירופרקטיקה קלינית, אתה יכול לשאול על XYMOGEN על ידי קורא 915-850-0900.
אקציטורעילות היא מנגנון פתולוגי הנראה במגוון בעיות בריאותיות שבהן עירור סינפטי מוגזם גורם למוות עצבי, וכן מאמינים כי נגרמת על ידי הצטברות חוץ-תאית של הנוירוטרנסמיטור המעורר גלוטמט, אשר מפעיל ומחבר יונוטרופי N-methyl-D-aspartate glutamatergic קולטנים (NMDARs) במוח. באופן כללי, NMDARs מווסתים ושומרים על סידן בתאים כדי לסייע בניהול מנגנונים פיזיולוגיים כמו פלסטיות סינפטית וזיכרון, עם זאת, גירוי מוגזם יכול בסופו של דבר להגביר את הסידן התוך תאי אשר מפעיל איתותים למוות תאי להפעלת אפופטוזיס. מנגנון פתולוגי זה הוצע במגוון נושאים בריאותיים, כגון פגיעה מוחית טראומטית (TBI) ומחלת אלצהיימר (AD), שם הוא נבדק בהרחבה כדי להבין בעיות בריאותיות וגישות טיפול. באירוע מוחי, הוכח כי רעקסיטוקסיות היא המנגנון הפתולוגי העיקרי שבו מתרחש נזק נוירוני והיא נחשבת למטרה ידועה עבור ניסיונות רבים לאחרונה לפיתוח תרופות לטיפול בשבץ מוחי. �
שבץ מוחי הוא בעיה חריפה בבריאות המוח אשר גורמת לנזק נוירוני אשר אין לה כיום גישות טיפול נוירו-פרוטקטיבי בטוחות ויעילות. מיד לאחר שבץ מוחי, רקמת המוח מאבדת זלוף דם ומרכז האוטם מתדרדר במהירות. זה גורם לאיסכמיה קלה יותר ותאי מוח או נוירונים רבים יגרמו למוות מושהה שיכול להימשך עד מספר שעות או אפילו ימים. מחקרים מראים כי מנגנון המוות של התא הוא בעיקר אקזיטוטוקסיות תלוית קולטן NMDA. באזורים איסכמיים, רמות הגלוטמט החוץ תאי עולות תוך מניעת שחרור גלוטמט, פעילות סינפטית או הפעלת NMDAR שהייתה מסוגלת להגביל את מוות התא במגוון מודלים של שבץ מוחי. לפיכך, מניעת רעילות אקסיטוטיות היא גישה טיפולית חשובה להפחתת נזק מוחי ושיפור מדדי תוצאות המטופלים לאחר שבץ מוחי, וזה בהחלט עודד מאמצים נרחבים לפיתוח גישות טיפול בשבץ מוחי מבוסס קולטן NMDA במהלך שני העשורים האחרונים. למרבה הצער, אלה נתקלו במידה רבה בתוצאות מאכזבות למדי. מספר מחקרים לא הצליחו למצוא את היעילות הצפויה של NMDAR להפחתת פציעות מוח. הסיבות מאחורי תוצאות המחקר הבסיסי והניסויים הקליניים עדיין לא ידועות, עם זאת, הוצעו מספר סיבות. אלה כוללים, בין היתר, את חוסר היכולת להשתמש במינונים הנכונים הנחוצים להגנה עצבית עקב תופעות הלוואי שלהם, חוסר היכולת להשתמש בתרופות בתוך חלונות ההגנה העצבית שלהן, עיצובים ניסויים לקויים והטרוגניות באוכלוסיית החולים. עם זאת, כפי שנסכם בקצרה במאמר הבא, שיפור בהבנתנו את המנגנונים הפיזיולוגיים והפתולוגיים של הפעלת NMDAR וכן את המסלולים השונים הקשורים לתתי סוגים שונים של NMDAR, אפשרו לחוקרים לפתח גישות טיפול חדשות המשפרות את החלונות הטיפוליים להגביר את הספציפיות עבור מסלולי איתות מוות, השגת הגנה עצבית מבלי להפריע למסלולי איתות חיוניים אחרים במורד הרצפטור של ה-NMDAR. �
נוגדי הגנה המכוונים לתת-סוגים של NMDAR
לתתי סוגים של NMDAR יש מטרות שונות באקסיטוטוקסיות ובפיזיולוגיה. ה-NMDAR הוא קולטן שיש לו בדרך כלל שתי יחידות משנה GluN1, המכונה גם NR1, וכן שתי יחידות משנה מתת-משפחת GluN2 (GluN2A-2D, הידועה גם בשם NR2A-2D). בקורטקס, תת-האוכלוסיות העיקריות של NMDARs הן קולטנים המכילים GluN2A או GluN2A ו-2B. קולטנים המכילים GluN2A נמצאים בסינפסות ואילו קולטנים המכילים GluN2B נמצאים על ממברנות חוץ-סינפטיות. קולטנים המכילים GluN2A ו-GluN2B שונים זה מזה מכיוון שהם מווסתים ומנהלים את הפלסטיות, ומעדיפים פוטנציציה ארוכת טווח (GluN2A) או דיכאון (GluN2B) באמצעות מגוון תכונות אלקטרופיזיולוגיות ותרופתיות כמו גם חלבוני איתות. בנוסף, קולטנים אלו ממלאים תפקיד מהותי בקידום הישרדות תאים (GluN2A) או מוות (GluN2B) לאחר גירוי אקציטוטוקסי. מכיוון שהקולטנים המכילים GluN2A מתמקדים בעיקר בסינפסות בעוד שהקולטנים המכילים GluN2B ממוקדים הן לממברנות הסינפטיות והן לממברנות החוץ-סינפטיות, כאשר מצבים אקציטוטוקסיים גורמים לגלוטמט להתרחב מעבר לסינפסות, איתות מוות בתיווך GluN2B מתחזק בהשוואה לאיתות הישרדות, אשר בסופו של דבר מביאים לכך. מוות. דרך שבץ, למשל, NMDARs נוטים פחות להעדיף הישרדות תאים ובמקום זאת יכולים לגרום להשפעות מזיקות על ידי מניעת מטרות פיזיולוגיות נורמליות ניכרות. Selfotel, חוסם NMDAR לא ספציפי, היה מגן על עצבים מפני שבץ במבחנה ובאינבו, אולם בסופו של דבר הוא לא הצליח להיות מגן על עצבים מפני שבץ בניסויים קליניים על ידי גרימת מגוון של תופעות לוואי בלתי נסבלות. �
אסטרטגיות טיפול להפחתת תופעות לוואי לא רצויות, כולל אנטגוניסטים לאתר גליצין ושיפורים ספציפיים לתת-סוג NMDAR, היו למקד את אזורי הקישור האלוסטריים של גליצין אלוסטריות ביחידות המשנה של GluN1 עם ליקוסטינל וגדסטין במקום לחסום ישירות את הקולטן. מועמדי תרופות אלו התמודדו היטב בבדיקות פרה-קליניות, אולם הם גם נכשלו כתוצאה מיעילות נמוכה למרות פרופיל תופעות לוואי מינימלי. תופעות הלוואי השליליות נבעו אולי מהחמצת חלון זמן בעקבות שבץ, שמראה אילו חוסמי קולטנים בטוחים ויעילים במניעת מוות. �
שיטות טיפול וטכניקות טובות יותר להפחתת תופעות לוואי לא רצויות של NMDAR הן לנצל את ההבדלים בין הווריאציות שלהן. לדוגמה, המעכב הספציפי ל-GluN2B traxoprodil מגן על עצבים במחקרי שבץ מוחי ותופעות לוואי מינימליות, עם זאת, הוא נכשל גם בניסויים קליניים. בדומה לאנטגוניסטים של אזור גליצין, ייתכן שהוא צריך להיות מווסת כראוי וניהולו לתפקד ביעילות. אגוניסטים של GluN2A צריכים לקדם איתות הישרדות תאים שיכול לאפשר התאוששות לאחר שבץ, כמו גם הישרדות תאים כדי למנוע איתות חולף. למען האמת, הפעלה של קולטנים המכילים GluN2A תוך שימוש במינונים מוגברים של גליצין הייתה מגנה עצבית במודל בעלי חיים של שבץ, אך מחקרים נוספים חייבים לבחון את הפעלת GluN2A כגישה טיפולית במשתתפים אנושיים. �
בעוד שאנטגוניסטים ומאפננים של NMDAR בטוחים ויעילים בהפחתת רעילות אקסיטוטיות בגרסאות ניסיוניות, חסרונם הוא האתגר ביישום גישות טיפול מוקדם כדי לחפף עם פסגת שחרור הגלוטמט האקציטוטוקסי. לחולי שבץ מוחי אין לעתים קרובות סיכוי לקבל גישות טיפול אלו בזמן. עם זאת, ניתן להימנע מהבעיה הבריאותית אם ניתן להשתמש בחוסמי קולטנים באוכלוסיות בסיכון. מחקר אחד הראה שמינונים נמוכים של ממנטין מניעתי, אנטגוניסט לא תחרותי של NMDAR עם מעט תופעות לוואי, יכולים להפחית במידה ניכרת את הפגיעה המוחית ואת הליקויים התפקודיים בעקבות שבץ מוחי. האם תרופות כלשהן נסבלות, בטוחות ויעילות כשהן נלקחות בדרך זו נותרה להוכחה, אך פתרונות חדשניים עשויים בכל זאת להתייחס לאופן מתן התרופות הללו. �
גורם אחד מלבד אלה של הניסויים הקליניים הכושלים הוא משחק הגומלין של NMDARs בהישרדות תאים שעלול להיות מובן לחלוטין לא נכון. בעשורים האחרונים, הצטברו עדויות לכך ש-NMDAR סינפטיים עלולים לגרום גם למוות של תאים ול-GluN2A, כמו גם ל-GluN2B, אין בהכרח פונקציות דיכוטומיות ב-exitotoxicity. מחקרים נוספים עשויים להידרש כדי להדגים אסטרטגיות מעכבי קולטן מגוונות יותר ולפתור מחלוקת זו. �
אמצעי הגנה נגד עצבים הממוקדים לאיתות מוות של תאים
גישת טיפול עבור מעכבי NMDAR היא להתמקד באירועים המורדים ביותר של מוות תאי המתרחשים על פני פרק זמן ארוך בהרבה לאחר הפעלת הקולטן. נקבעו מגוון מסלולי מוות של תאים בעקבות הפעלה ומספר קבוצות סיפקו הוכחה עקרונית לכך שניתן לווסת מסלולים אלו ולנהל אותם באמצעות שימוש בפפטידים כדי להגן בסופו של דבר על תאי מוח או נוירונים ללא כל תופעות לוואי. �
אסטרטגיית הפפטידים הוותיקה ביותר המדווחת והנחקרת ביותר ביעדי שבץ היא מוות תאי בתיווך תחמוצת החנקן סינתאז (nNOS). NNOS מתחבר לחלבון פוסט-סינפטי 95 (PSD95) אשר מתחבר לאחר מכן לזנב C-terminal של תת-היחידה GluN2B. NOS הוא אנזים מופעל בסידן אשר מפעיל את התפתחות תחמוצת החנקן (NO) ואת מצבו שלו בקומפלקס הקולטנים אשר מקשר אותו בסמיכות לזרם הממוקד של סידן הנכנס ל- GluN2B המופעל. באירוע מוחי, זרימת הסידן המופרזת מפעילה את nNOS המזוהה עם GluN2B. פפטיד הפרעה משמש לניתוק הקומפלקס כדי למנוע התפתחות NO. הפפטיד, Tat-NR2B9c, מורכב מרצף חדירת תאים שמקורו ב-HIV-1 Tat המאפשר מעבר דרך מחסום הדם-מוח וממברנות התא, המחוברים לעותק של האזור ב-GluN2B עבור PSD95. הפפטיד ו-GluN2B מנתקים את PSD95, לפיכך, מנתקים את ה-nNOS ברמות הניכרות המקומיות של סידן מבלי להפריע לתפקוד הקולטן ממסלולים שונים. ניצול מביא להגנה ניכרת מפני נזקים לרקמות ולנזקים תפקודיים ללא תופעות לוואי in vitro ו-in vivo לאחר מנה בודדת שניתנה לפני או אחרי איסכמיה in vivo. הפפטיד הצליח לאחרונה בניסוי קליני שלב II שבו הפחית אוטמים יאטרוגניים במהלך טיפול במפרצת תוך גולגולתית. זוהי הפעם הראשונה שמחקר מחקר הוכיח יעילות בבני אדם, אשר גם מראה אותנטיות כי מיקוד מוות תאי במורד הזרם יכול להועיל נגד פציעות עצביות אקזיטוטוקסיות/איסכמיות. �
בעוד שהשימוש בפפטידים בסביבה קלינית בטוח ויעיל, יעילות דומה הושגה עם תרופות מולקולות קטנות הפועלות בדיוק על אותה מטרה ומתפקדות כמו הפפטידים במעבדה. כדי לחקות את Tat-NR2B9c, הוכחו בנפרד שתי מולקולות קטנות, IC87201 ו-ZL006 מתחרות באזור החיבור הספציפי הזהה ל-GluN2B מבלי להשפיע על החיבור של PSD95 לחלבונים אחרים. בנוסף, ZL006 מחקה את ההגנה העצבית של הפפטיד מבלי לגרום לתופעות לוואי משמעותיות. על ידי זיהוי המטרות והאזורים הספציפיים, מחקרים יכולים לדמות תרופות מולקולות קטנות ולהאיץ את גילוין לקראת רעילות ושבץ מוחי. �
מסלולים אחרים הספציפיים ל-GluN2B הוכחו באופן דומה והם מראים הבטחה בשלבי הפיתוח. מסלול אחד כזה המופעל בעקבות הפעלת GluN2B הוא העצמה וגיוס של GluN2B בממברנת התא על ידי חלבון קינאז 1 (DAPK1) הקשור למוות. DAPK1 הוא חלבון שמתחבר לקלמודולין כדי להפעיל אפופטוזיס אך הוא מזורחן בצורה לא פעילה שאינה מסוגלת לקשר בין מוות תאי לבין קלמודולין. בעקבות אקציטורעילות, הפעלת קלציניורין משחררת ומעוררת DAPK1, תורמת למוות של תאים. יתר על כן, DAPK1 פעיל יכול להתחבר לזרחן לזנב C-טרמינלי של קולטנים, אקזיטוטוקסיות ותפקודם, להחמיר את זרימת הסידן. פפטיד הפרעה מקושר ל-Tat שיש לו את אזור הזרחון C-tail שהוא GluN2B הצליח לחסום את האינטראקציה של DAPK1 פעיל עם GluN2B ולקדם רעילות אקזיטוטיות. ברגע שהפפטיד נוצל בעכברים, שכונו Tat-NR2B-CT, הוא שיפר את התוצאה בעקבות איסכמיה. עם זאת, Tat-NR2B-CT היה יעיל רק במניעת פעילות והחדרה בורחת במקום האפופטוטי במורד הזרם של איתות DAPK1. החוקרים הצליחו גם לחבר ולהנחות את DAPK1 לעבר ליזוזומים על ידי הכללת רצף בסגירת הפפטיד המפריע ליצירת פפטיד פירוק. התוצאה הייתה ירידה רצינית וזמנית ברמות DAPK1 העמוסות עם ירידה מקבילה באוטם בעת מתן הפפטיד שעות לאחר איסכמיה, על פי מספר מחקרים. �
ה-c-Jun N-terminal kinase 3 (JNK) פועל על מסלולים רבים ומהווה מתווך למוות תאי באקסיטוטוקסיות. JNK interacting protein (JIP) מחבר ומונע פעילות JNK באמצעות תחום מקשר JNK (JBD) המשתרע על פני 20 שיירים. כאשר שיירים אלו מחוברים ל-Tat החל מהפפטיד המופרע של Tat-JBD20, הם מסוגלים להגביל את פעילות JNK ולמנוע מוות של תאים במודלים של שבץ כאשר הם ניתנים לפני או אחרי איסכמיה. הוכח כי הפפטיד Tat-JBD20 משתמש בחומצות אמינו D במקום חומצות אמינו L כדי לעמוד בפירוק על ידי פרוטאזות אנדוגניות. פעולה זו מגדילה מאוד את זמן מחצית החיים של הפפטיד ואינה משפיעה לרעה על זיקת הקישור והסלקטיביות שלו, מה שמוכיח שניתן לנצל את השינוי הזה עבור מספר פפטידי הפרעה כדי להגביר את היעילות והזמינות הביולוגית. �
מטרות חדשות מתגלות תמיד. בעוד שכרגע, לא נעשה שימוש בגישות חדשות לטיפול בשבץ מוחי, התקדמות רבה הושגה על ידי מיקוד התהליכים המתרחשים במהלך שבץ ליצירת גישות טיפול. עם הופעת הבכורה של השגת פפטידי השפלה והפרעה המכוונים לאירועי איתות חולפים ספציפיים ל-GluN2B, יש תקווה שטיפולים חדשים נמצאים באופק לבעיות בריאותיות שיש להן רעילות מעוררת. �
אקסיטוטוקסיות היא המנגנון הפתולוגי שבאמצעותו נפגעים או מונעים בסופו של דבר תאי מוח או נוירונים על ידי גירוי מופרז ממועברים עצביים, כולל גלוטמט וחומרים דומים אחרים. זה מתרחש בסופו של דבר כאשר קולטני ה- NMDA וקולטני ה- AMPA מופעלים מדי על ידי קולטני גלוטמט מוליכים עצביים. זה יכול לגרום למגוון תהליכים העלולים לפגוע במבני התא, כולל רכיבים של שלד הציטוס, הממברנה ו- DNA. ויסות וניהול רעילות מרגשות יכול לעזור לשמור על רווחת הכלל. - ד"ר אלכס Jimenez DC, תובנה CCST
אקסיטוטוקסיות היא מנגנון פתולוגי בו עירור סינפטי מופרז גורם למוות עצבי ונחשבים כי הוא גם נגרם כתוצאה מהצטברות חוץ-תאית של הגלוטמט המרגש העצבתי המעורר ומחבר בין קולטנים N-methyl-D-aspartate קולטנים glutamatergic (NMDARs) במוח . מנגנון פתולוגי זה הוצע במגוון סוגיות בריאותיות, כמו פגיעה מוחית טראומטית (TBI) ומחלת אלצהיימר (AD), שם הוא נבדק בהרחבה להבנת סוגיות בריאותיות וגישות טיפול. היקף המידע שלנו מוגבל לנושאי כירופרקטיקה, שרירים ושלד עצבים וכן מאמרים, נושאים ודיונים ברפואה תפקודית. לדיון נוסף בנושא לעיל, אנא אל תהסס לשאול את ד"ר אלכס חימנז או צור איתנו קשר בכתובת 915-850-0900 .
אוצר על ידי ד"ר אלכס חימנז
הפניות
לי, ויקטור ויו טיאן וואנג. � מנגנונים מולקולריים של אקזיטוטוקסיות בתיווך קולטן NMDA: השלכות על טיפולים נוירו-פרוטקטיביים לשבץ מוחי.� מחקר התחדשות עצבית, Medknow Publications & Media Pvt Ltd, נובמבר 2016, www.ncbi.nlm.nih.gov/pmc/articles/PMC5204222/.
דיון בנושא נוסף: כאב כרוני
כאב פתאומי הוא תגובה טבעית של מערכת העצבים המסייעת להפגנת פגיעה אפשרית. דרך אגב, אותות כאב נודדים מאזור פצוע דרך העצבים וחוט השדרה למוח. בדרך כלל הכאב פחות חמור ככל שהפציעה מחלימה, עם זאת, כאבים כרוניים שונים מהסוג הממוצע של הכאב. עם כאבים כרוניים, גוף האדם ימשיך לשלוח אותות כאב למוח, ללא קשר אם הפגיעה נרפאה. כאב כרוני יכול להימשך מספר שבועות ואף מספר שנים. כאבים כרוניים יכולים להשפיע בצורה אדירה על ניידות המטופל והוא יכול להפחית את הגמישות, הכוח והסיבולת.
צוהר עצבי פלוס למחלות נוירולוגיות
ד"ר אלכס חימנז משתמש בסדרת בדיקות המסייעות להערכת מחלות נוירולוגיות. הזומר העצביTM בנוסף הוא מערך של נוגדנים אוטומטיים נוירולוגיים המציע זיהוי נוגדנים-לאנטיגן ספציפי. הזומר העצבי התוססTM פלוס נועד להעריך את תגובתו של האדם ל- 48 אנטיגנים נוירולוגיים עם קשר למגוון מחלות הקשורות לנוירולוגיה. הזום העצבני התוססTM Plus שואפת להפחית מצבים נוירולוגיים על ידי העצמת חולים ורופאים עם משאב חיוני לגילוי סיכונים מוקדם והתמקדות משופרת במניעה ראשונית בהתאמה אישית.
נוסחאות לתמיכה במתילציה
XYMOGEN s נוסחאות מקצועיות בלעדיות זמינות באמצעות אנשי מקצוע נבחרים בתחום הבריאות. מכירה והנחה באינטרנט של נוסחאות XYMOGEN אסורות בהחלט.
בגאווה, ד"ר אלכסנדר חימנז עושה XYMOGEN נוסחאות זמין רק לחולים תחת הטיפול שלנו.
נא להתקשר למשרד שלנו על מנת שנוכל להקצות רופא התייעצות לגישה מיידית.
אם אתה חולה של פגיעה רפואית וכירופרקטיקה קלינית, אתה יכול לשאול על XYMOGEN על ידי קורא 915-850-0900.
הכלי Find A Practitioner של IFM הוא רשת ההפניות הגדולה ביותר ברפואה פונקציונלית, שנוצרה במטרה לסייע למטופלים לאתר מטפלים ברפואה פונקציונלית בכל מקום בעולם. מטפלים מוסמכים של IFM מפורטים ראשונים בתוצאות החיפוש, לאור השכלתם הרחבה ברפואה פונקציונלית